
La fibrosi cistica (FC) è una malattia genetica degenerativa multiorgano causata da mutazioni a carico del gene CFTR (cystic fibrosis transmembrane conductance regulator) situato sul braccio lungo del cromosoma 7.
Mutazioni del CFTR determinano la produzione di una proteina canale difettosa nella sua funzione o addirittura ne impediscono la sintesi. In assenza di un canale CFTR funzionante, il cloro e il bicarbonato non possono muoversi dall’interno delle cellule epiteliali verso l’esterno.
La mancata fuoriuscita dello ione cloro e dello ione bicarbonato dalle cellule causa un riassorbimento di acqua e sodio che disidrata i liquidi all’esterno, rendendo il muco che riveste internamente gli organi denso e vischioso. Da questo caratteristico muco “viscido” deriva il nome con cui l’FC era nota in passato: mucoviscidosi.
Quali sono i sintomi della Fibrosi Cistica?
Le secrezioni dense, tipiche della FC, danneggiano soprattutto l’apparato digestivo e quello respiratorio. Una prima manifestazione della patologia è l’ileo da meconio, un’occlusione intestinale che si verifica quando il meconio (le prime feci espulse dal neonato) è estremamente spesso e colloso, tanto che il neonato non riesce ad evacuare spontaneamente.
Nei polmoni, il muco denso ostruisce le vie aeree, intrappolando al suo interno batteri ed altri microrganismi che provocano infezioni persistenti e infiammazione cronica, con una progressiva insufficienza respiratoria.
Poiché questi microrganismi (non in grado di causare malattia nei soggetti sani), possono essere trasmessi da una persona FC ad un’altra (cross-infection), ai pazienti è fortemente sconsigliato socializzare ed è raccomandato l’utilizzo di mascherine e il mantenimento di una distanza minima di 2 metri ogni qualvolta si trovino in presenza di altri pazienti FC o di persone non FC potenzialmente contagiose.
Leggi anche: La Fibrosi Polmonare – Di cosa si tratta
Sintomi polmonari comuni ai pazienti FC sono tosse persistente, bronchiti e polmoniti e, nelle fasi più avanzate di malattia, fiato corto. Nell’85% dei malati FC, è presente un’insufficienza pancreatica causata dall’accumulo di muco nel pancreas che impedisce agli enzimi pancreatici di raggiungere l’intestino e partecipare all’assorbimento di grassi e altri nutrienti.
Ne consegue un difetto di digestione dei cibi, diarrea (per quanto negli adulti siano anche possibili ostruzioni intestinali), malassorbimento, ritardo di crescita nel bambino e scadente stato nutrizionale nell’adulto.
Inoltre, le secrezioni stagnanti possono formare delle specie di cisti che vengono circondate da tessuto fibrotico: da questa manifestazione deriva il nome “fibrosi cistica” (nome completo “fibrosi cistica del pancreas”). Con l’età, il progredire del danno pancreatico può causare una forma di diabete (Diabete correlato alla Fibrosi Cistica) con caratteristiche comuni sia al diabete tipo 2 sia al diabete tipo 1. Nel fegato, il muco denso ostruisce i dotti biliari, una situazione che può portare a epatopatia e cirrosi epatica.
In quasi tutti gli uomini FC, la mancanza di dotti deferenti causa infertilità maschile da assenza di spermatozoi nel liquido seminale (azoospermia) e l’impossibilità di concepire figli se non attraverso tecniche di fecondazione assistita.
Infine, a causa dell’anomalo funzionamento della proteina CFTR che concentra il sale a livello della cute, la pelle dei pazienti FC ha la “curiosa” caratteristica di risultare salata. Questa caratteristica era nota già nel diciassettesimo secolo, come testimonia un antico proverbio tedesco che così recitava: “Morirà presto il bambino la cui fronte sa di sale se baciata”, anticipando l’utilizzo del sudore salato dei pazienti come test diagnostico di fibrosi cistica.

La genetica delle Fibrosi Cistica
A poco più di 30 anni dall’identificazione della prima mutazione del gene CFTR (1989), sono state individuate ormai più di 2.000 varianti (non tutte responsabili di malattia), divise in 6 classi in base al meccanismo di alterazione del funzionamento della proteina CFTR.
I soggetti con FC ereditano due copie mutate del gene CFTR, una da ogni genitore. Questi ultimi sono “portatori sani” e hanno una sola copia difettiva del gene CFTR, non sufficiente a determinare la manifestazione della malattia.
In genetica, una malattia che si trasmette secondo questa modalità è detta “autosomica recessiva”. La frequenza di portatori sani in Italia è di 1 ogni 25 persone circa. In altre parole, circa una coppia su 600 è composta da due portatori che, ad ogni gravidanza, ha:
- il 25% di probabilità di avere un bambino affetto da fibrosi cistica, a cui entrambi i genitori trasmettono la copia difettiva del gene;
- il 50% di probabilità di avere un figlio portatore sano, a cui un genitore trasmette la copia sana e l’altro la copia difettiva del gene;
- il 25% di probabilità di avere un figlio sano, non portatore, a cui entrambi i genitori trasmettono la copia sana del gene.
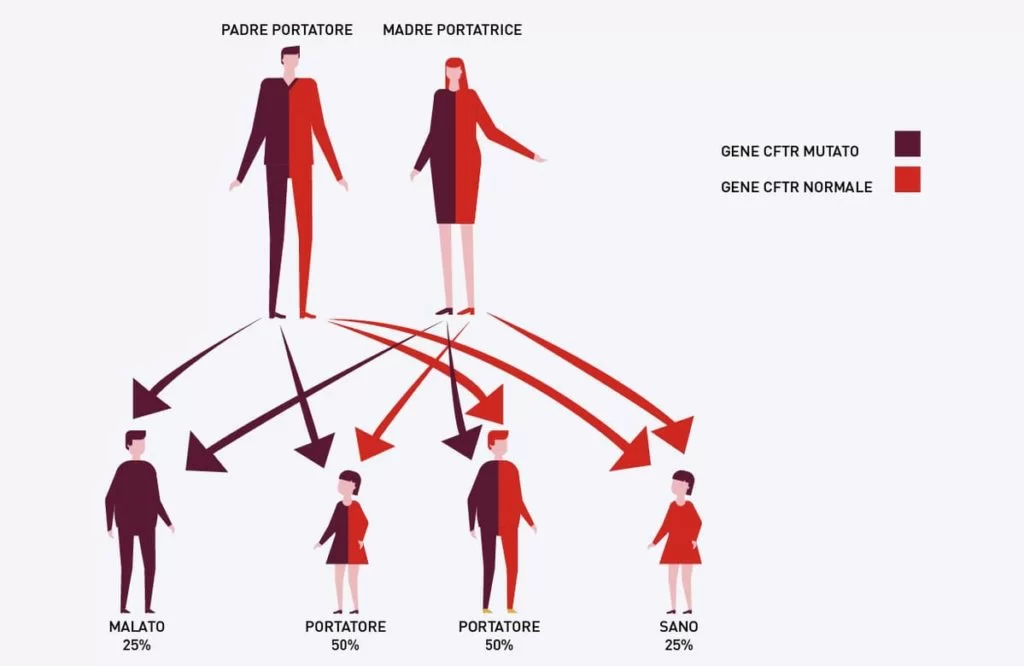
I soggetti FC, a loro volta, trasmettono le copie mutate del gene ai loro figli. Nel caso in cui il partner sia un portatore sano, la probabilità di avere un figlio con FC è del 50%. Nell’altra metà dei casi, il figlio sarà un portatore sano della malattia.
L’alta frequenza di portatori sani e di coppie di portatori nella popolazione è la ragione per cui mediamente un bambino ogni 2.500-3.000 nuovi nati avrà l’FC, la più frequente fra le malattie genetiche gravi, che colpisce circa 70.000 persone nel mondo.
Come si diagnostica la Fibrosi Cistica?
La diagnosi di fibrosi cistica è un processo multistep, che si basa su tre elementi fondamentali:
- screening neonatale positivo o un quadro clinico compatibile con la malattia;
- test del sudore positivo (a conferma di una disfunzione del canale CFTR);
- l’identificazione di due mutazioni che possono causare la malattia attraverso il test genetico.
Nello screening neonatale si esegue il dosaggio della tripsina (un enzima pancreatico) su una goccia di sangue del bambino prelevato a poche ore dalla nascita; un primo test positivo richiede un secondo dosaggio della tripsina tra il 15°e il 20° giorno di vita del neonato. In caso di tripsina elevata la diagnosi di FC va confermata o esclusa ricorrendo ad altri test.
Il test del sudore, in grado di diagnosticare circa il 90% dei casi di malattia, è l’esame di riferimento. Si esegue dosando il cloro presente nel sudore raccolto in una garza dopo aver stimolato le ghiandole sudoripare tramite applicazione di un’apposita sostanza (pilocarpina) e di un bassissimo stimolo elettrico (iontoforesi).
La conferma diagnostica richiede sempre la ricerca delle mutazioni del gene CFTR (test genetico), esame che diventa dirimente nei pazienti con test del sudore border-line oppure in casi con test del sudore negativo, ma sintomi clinici sospetti.
Il test genetico di primo livello consiste nella ricerca delle più frequenti mutazioni che causano la FC nella popolazione di appartenenza dell’individuo e consente di individuare oltre l’80% dei bambini affetti.
Solo in casi selezionati è indicato lo studio completo del gene CFTR (2.200 mutazioni oggi identificate) attraverso indagini molecolari come il sequenziamento e lo studio delle delezioni.

Quale terapia per la Fibrosi Cistica?
La fibrosi cistica è una malattia per cui ancora oggi non esiste cura, ma per cui sono a disposizione trattamenti terapeutici di vecchia e nuova generazione. Le scopo delle terapie di vecchia generazione è di controllare la malattia in tutte le sue manifestazioni e prevenire le complicanze secondo protocolli terapeutici condivisi a livello internazionale.
Queste terapie “sintomatiche” prevedono antibioticoterapia per via aerosolica o sistemica per trattare le infezioni polmonari, aerosolterapia e fisioterapia respiratoria per rimuovere il muco denso e vischioso e ridurre l’infiammazione polmonare, terapia enzimatica sostitutiva e approccio nutrizionale per migliorare lo stato nutrizionale, ma anche attività fisica e sportiva, sostegno psicologico e sociale. In caso di malattia molto avanzata, il trapianto polmonare può rappresentare una prospettiva terapeutica.
I trattamenti terapeutici di nuova generazione nascono dalla conoscenza dei meccanismi attraverso cui le diverse mutazioni del gene CFTR alterano la funzione della proteina canale e sono basati sull’utilizzo di “modulatori” che agiscono regolando la funzione della proteina CFTR mutata (potenziatori) e/o correggendo i suoi spostamenti all’interno della cellula (correttori).
Queste nuove terapie, specificatamente disegnate (personalizzate) per contrastare l’effetto di specifiche mutazioni o classi di mutazioni, sono però destinate esclusivamente ai pazienti che presentano tali varianti genetiche.
Leggi anche: L’EMA raccomanda l’approvazione di Kaftrio per la Fibrosi Cistica

Accanto ai 4 modulatori già esistenti (Ivacaftor, Lumacaftor,Tezacaftor, Elexacaftor) che hanno mostrato il potenziale di modificare sensibilmente il decorso della malattia, molti altri principi attivi sono in fase di sperimentazione preclinica, con lo scopo di offrire questa “terapia personalizzata” ad un più ampio numero di pazienti.
I modulatori però, non correggendo il difetto genetico di base, non possono essere considerati la cura definitiva della FC; per questo i pazienti devono comunque continuare a seguire la terapia di vecchia generazione di prevenzione e cura delle complicanze.
Al contrario, la correzione definitiva del gene CFTR (terapia genica) è stata tentata negli anni utilizzando vettori virali per inserire nelle cellule polmonari la sequenza corretta del gene CFTR. Questa strategia ha ottenuto un’espressione a bassi livelli e non permanente del gene CFTR a causa, tra gli altri fattori, del rapido turnover delle cellule epiteliali polmonari, della risposta immunitaria associata all’utilizzo di alcuni virus e della loro breve persistenza nelle cellule polmonari che richiedeva trattamenti ripetuti.
Più recentemente, con approccio di editing genetico basato su CRISPR-Cas (un complesso di proteine che funziona da “forbice molecolare” e da “correttore” di sequenze di DNA), si è ottenuta una correzione permanente del difetto genetico e un completo recupero di un canale CFTR funzionante in modelli ex vivo (cellule prelevate da pazienti).
Prospettive future
La fibrosi cistica è una malattia complessa, con tipologia e severità dei sintomi che varia da persona a persona, il cui decorso risente di numerosi fattori, prima fra tutti l’età della diagnosi: la diffusione dello screening neonatale a livello nazionale ha permesso di abbassare notevolmente l’età media alla diagnosi con risvolti prognostici positivi.
La diagnosi precoce permette infatti di ritardare il più possibile l’evoluzione della malattia, attuando da subito schemi terapeutici volti a prevenire complicanze e danni irreversibili di organi ed apparati.
I progressi notevoli nella cura specializzata e multidisciplinare per la FC hanno migliorato le condizioni e l’aspettativa di vita dei pazienti: mentre negli anni 50 un bambino con l’FC raramente viveva abbastanza a lungo da frequentare la scuola elementare, oggi i pazienti con FC frequentano le università, intraprendono carriere, si sposano e hanno figli.
Nonostante l’aspettativa di vita sia migliorata, l’evoluzione del danno a livello del polmone FC rappresenta ancora la principale causa di morbilità e mortalità e, in caso di malattia molto avanzata, il trapianto bipolmonare rimane l’unica terapia possibile.
Leggi anche: Fibrosi Cistica, il punto dell’Ospedale Bambin Gesù