
 La malattia di Tay-Sachs, patologia rara del sistema nervoso tramandata ereditariamente, è dovuta alla mancanza, nel sangue del bambino, di certi enzimi che normalmente intervengono nel metabolismo dei lipidi. Il morbo di Tay-Sachs appartiene al gruppo delle gangliosidosi, patologie da accumulo lisosomiale causate dalla carenza di enzimi attivi nei lisosomi (organuli cellulari deputati alla degradazione di determinate molecole), con conseguente accumulo delle molecole non degradate. Solitamente letale, il morbo colpisce i lattanti ed in genere è associato a cecità e ritardo mentale.
La malattia di Tay-Sachs, patologia rara del sistema nervoso tramandata ereditariamente, è dovuta alla mancanza, nel sangue del bambino, di certi enzimi che normalmente intervengono nel metabolismo dei lipidi. Il morbo di Tay-Sachs appartiene al gruppo delle gangliosidosi, patologie da accumulo lisosomiale causate dalla carenza di enzimi attivi nei lisosomi (organuli cellulari deputati alla degradazione di determinate molecole), con conseguente accumulo delle molecole non degradate. Solitamente letale, il morbo colpisce i lattanti ed in genere è associato a cecità e ritardo mentale.
Malattia di Tay-Sachs e cause, la genetica
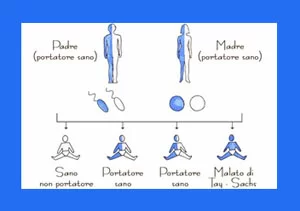
La malattia di Tay-Sachs è un disordine autosomico recessivo; ciò significa che se entrambi i genitori sono portatori, vi è un rischio del 25% di dare alla luce un figlio affetto. Per potersi ammalare quindi, il bambino deve ricevere due copie del gene difettoso (in questo caso situato sul cromosoma 15), una da ogni genitore. Se solo un genitore trasmette il gene difettoso, il bambino è chiamato vettore: non svilupperà la malattia, ma avrà la possibilità di trasmetterla ai propri figli. Chiunque può essere un vettore di Tay-Sachs, ma la malattia è più comune tra la popolazione ebrea Ashkenazi (circa un membro della comunità ashkenazita ogni 27 trasporta il gene della malattia di TS).
Il morbo di Tay-Sachs è caratterizzato dall’accumulo di quantità nocive, in particolare nelle cellule neurali, di una sostanza chiamata ganglioside GM2. Questa sostanza viene accumulata perché il corpo non produce un enzima (proteina), chiamato esosaminidasi, responsabile del metabolismo del ganglioside GM2. A causa di questo difetto genetico del metabolismo, il lattante non è in grado di utilizzare certe sostanze grasse presenti nei cibi che finiscono quindi per accumularsi nel cervello, provocando la distruzione delle cellule cerebrali.
Quante forme di Morbo di Sachs esistono?
In base all’età di esordio ed alla sintomatologia si possono distinguere tre forme:
- forma infantile o classica: compare tra i 3 e i 6 mesi di vita e si manifesta nel bambino con ritardo psicomotorio, ipotonia e in alcuni casi megalocefalia (aumento delle dimensioni del cranio). Nel giro di pochi mesi i bambini colpiti smettono di sviluppare nuove abilità e perdono quelle già acquisite. Possono comparire crisi convulsive e, poco a poco, si assiste ad una diminuzione dei movimenti volontari e della vista; la debolezza muscolare è progressiva ed esita in paralisi. Il segno più precoce è rappresentato dagli scatti improvvisi e continui in risposta al rumore;
- forma giovanile (tipo 2): l’esordio avviene tra i 2 ed i 6 anni di vita con difficoltà nella deambulazione. I sintomi sono disturbi del comportamento, atassia (mancanza di coordinazione dei movimenti muscolari volontari) e regressione mentale;
- forma dell’adulto o cronica (tipo 3): può esordire attorno ai 10 anni di vita ma spesso la diagnosi è possibile solo in età adulta. Prevalgono i sintomi neurologici quali difficoltà a camminare, a controllare i movimenti e a parlare. Possono essere presenti anche manifestazioni psichiatriche.
Il decorso della malattia è in genere molto rapido e la prognosi è decisamente negativa: il bambino muore di solito intorno ai 4 o 5 anni. L’insorgenza tardiva in età adulta è molto rara.
Sintomi della malattia di Tay-Sachs
Il lattante si sviluppa normalmente fino all’età di circa 3-6 mesi, età in cui iniziano a manifestarsi i sintomi associati alla malattia di Tay-Sachs:
- sordità
- cecità
- crescita lenta
- inabilità nel deglutire
- competenze mentali ritardate
- attacchi epilettici
- demenza
- perdita di forza
- incapacità di sorridere.
La diagnosi
 La diagnosi si basa sulle manifestazioni cliniche e sulla misurazione dell’attività dell’enzima esosaminidasi A. Gli esami volti a diagnosticare la malattia di TS sono l’analisi degli enzimi del sangue e la visita oculistica (rivela una macchia rosso ciliegia nella macula). Nelle famiglie a rischio è possibile effettuare diagnosi prenatale mediante analisi genetica, in seguito ad amniocentesi o villocentesi.
La diagnosi si basa sulle manifestazioni cliniche e sulla misurazione dell’attività dell’enzima esosaminidasi A. Gli esami volti a diagnosticare la malattia di TS sono l’analisi degli enzimi del sangue e la visita oculistica (rivela una macchia rosso ciliegia nella macula). Nelle famiglie a rischio è possibile effettuare diagnosi prenatale mediante analisi genetica, in seguito ad amniocentesi o villocentesi.
Lo screening prenatale nel morbo di Sachs
L’origine etnica e la storia familiare sono indicatori del bisogno di consulenza genetica per evitare di passare la mutazione al proprio figlio. Lo screening prenatale è fondamentale per individuare gli individui a rischio e consente di mettere in pratica, il prima possibile, tutti i possibili provvedimenti. Lo screening si effettua mediante un semplice prelievo del sangue volto ad analizzare l’attività della beta-esosaminidasi A; è disponibile anche un prelievo per l’esame del DNA genetico che analizza la mutazione genetica a carico dell’enzima. Se entrambi i genitori sono portatori sani di Tay-Sachs sono disponibili tecniche di riproduzione assistita che combinano la diagnosi genetica pre-impianto con la fecondazione in-vitro, al fine di aiutare i genitori a prevenire la trasmissione di questo disturbo genetico.
Terapia della malattia di Sachs
Purtroppo oggi non esiste ancora un trattamento per la malattia di Tay-Sachs, ma solo modi per alleviare le sofferenze dei piccoli che ne sono affetti. In genere vengono prescritti farmaci antiepilettici per tenere sotto controllo le crisi convulsive mentre per il trattamento delle forme progressive è in fase di studio una terapia in grado di inibire la sintesi dei gangliosidi.
La storia di Ludovica
“Rassegnatevi, per vostra figlia non ci sono cure, le restano uno o due anni di vita. E’ inutile portarla in giro per ospedali”. Roberta, racconta come se fosse ieri il giorno della diagnosi fatta a sua figlia Ludovica: malattia di Tay-Sachs di tipo giovanile, questo il verdetto dopo mesi di analisi. Contrariamente al normale decorso della malattia (la prognosi era di uno, massimo due anni di vita) Ludovica, che aveva perso qualsiasi contatto cognitivo, ha ricominciato a dire alcune parole, si muove un po’ meglio, ha più forza e gli elettroencefalogrammi mostrano un deciso miglioramento. Tutto questo grazie alla tenacia della sua famiglia che non si è rassegnata e al Dottor Bruno Bembi di Udine, che ha deciso di sottoporla ad una terapia sperimentale fuori protocollo: il Miglustat. Il Dottor Bembi, che da anni studia questo tipo di malattie, ha attivato una terapia sperimentale che sembra riuscire ad arrestare la terapia; al momento si tratta ancora di un “farmaco orfano”, prodotto cioè per un numero esiguo di pazienti. Nonostante il farmaco rallenti la progressione della malattia, è importante non farsi false speranze: il Miglustat, come affermato dallo stesso Dottor Bembi, non rappresenta la cura di questa rara patologia.
Mamma Roberta però non si arrende e continua a sperare che, diffondendo la storia di sua figlia, altri ricercatori studino la malattia così da poter aiutare Ludovica e, un domani, altri bambini come lei.